Genética
Curso: Odontologia Universidade Federal de Juiz de Fora
Período: 2° / 99
Autores:
- Ciro Reis Rodrigues
- Eduardo Mattos Ponce de Lion
- Frederico Kleinsorge Daibert
- Gustavo Silva Gouvêa
- José Fontes
- Leonardo Pereira Lucas
Tópicos
• INTRODUÇÃO
• AGENESIA DENTÁRIA
• DOENÇAS HEREDITÁRIAS QUE AFETAM OS DENTES
• MANIFESTACÕES ORAIS E FACIAIS DAS ANOMALIAS CITOGENÉTICAS
• DOENÇAS CROMOSSÔMICAS
• SÍNDROME DE TURNER (45,X e variantes)
• TRISSOMIA DO X (47,XXX)
• SÍNDROME DE
KLINEFELTER (47,XXY)
• SÍNDROME 47,XYY
• SÍNDROME DO MIADO DE GATO (5p-)
• ODONTOLOGIA RACIAL
• AÇÃO GÊNICA
• ANÁLISE GENEALÓGICA
• CONCLUSÃO
• GLOSSÁRIO
INTRODUÇÃO
Tudo o que somos: a nossa aparência, os traços da personalidade, a
maneira como reagimos nas relações com o mundo físico e outros seres,
diferentes ou semelhantes, é o resultado de uma complexa interação, a nível
molecular, celular e de organismo, entre o material biológico que herdamos de
nossos genitores e o meio ambiente.
Há duas filosofias básicas a adotar com relação às ocorrências que
alguém pode encontrar em sua vida: a resignação e a adaptação, ou a rebeldia e
tentativa de mudança. É muito provável que haja um substrato genético (mediado
por hormônios e outras substâncias) que irá condicionar, em parte, qual das
duas atitudes será tomada por uma determinada pessoa, embora o ambiente físico
e sócio-cultural, bem como a história individual, também possam influir.
Ambas as filosofias estiveram bem representadas, durante os primeiros
anos deste século, entre os geneticistas e pesquisadores de áreas afins. O
título de um livro sobre herança publicado em 1974, por Rife, Dados do destino, pode ser mencionado
como exemplo para o primeiro tipo de atitude. A decisão sobre ter ou não ter
uma doença genética estaria fora do nosso controle. Associada a esta, existia a
idéia, arraigada até hoje, de que moléstia hereditária não tem cura.
O movimento eugenista da década dos 30 pode ser indicado como
representativo do segundo tipo de filosofia. Havia a crença ingênua de que,
através de um trabalho vigoroso, seria possível eliminar-se males sociais ou problemas
físicos geneticamente condicionados num período relativamente curto de tempo. A
extensão dessas idéias e sua deturpação através da ''higiene racial' 'da
Alemanha nazista, desenvolvida especialmente na década seguinte, trouxe como
conseqüência o afastamento dos geneticistas conscientes do movimento eugênico;
e a conotação negativa que tal termo criou foi tamanha que as duas sociedades
com esse nome existentes nos Estados Unidos e na Inglaterra resolveram mudar o
título das suas revistas, substituindo a palavra "eugenia" por
"biologia social" ou "ciência bio-social".
Progressos recentes no conhecimento da genética e ciências afins estão
possibilitando um controle cada vez mais estrito sobre a reprodução humana, com
o poder de influência direta sobre o DNA ; por outro lado, o número de doenças
genéticas passíveis de diversos tipos de tratamento aumentou consideravelmente
nos últimos anos, tornando obsoleta a idéia de que nada se poderia fazer nesses
casos.
AGENESIA DENTÁRIA
Situações de consultório
A mãe de uma criança vem consultá-lo informando que nunca se
desenvolveram dentes em seu filho, apesar de já estar com 6 anos de idade. Ela
inveja sua vizinha, que teve uma criança que já nasceu com dois incisivos
centrais mandibulares à mostra. Na semana seguinte uma jovem de 13 anos,
preocupada com sua aparência, procura-o para que lhe sejam colocados incisivos
laterais maxilares artificiais, pois o do lado esquerdo é muito reduzido, e o
do lado direito nunca apareceu. Outro jovem, totalmente calvo apesar de ter
apenas 22 anos de idade e apresentando dificuldades de transpiração vem colocar
uma dentadura postiça, devido à ausência (não erupção) de l0 dentes. Nos dois
últimos casos há outras pessoas similarmente afetadas nas famílias dos mesmos.
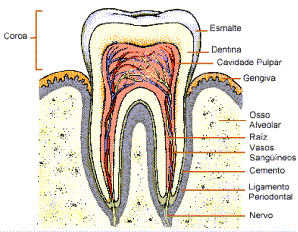
Todas essas situações relacionam-se com graus diversos de AGENESIA DENTÁRIA, que pode expressar-se desde a ANODONTIA (ausência total de dentes) até a HIPODONTIA (ausência de um ou poucos dentes).
Para compreender perfeitamente os casos acima apresentados, é necessário, preliminarmente, uma análise:
(a) Do processo de divisão celular que ocorre nos tecidos do embrião em desenvolvimento e continua em muitos órgãos do indivíduo adulto, a MITOSE, Divisões desiguais na quantidade de citoplasma destinado às células-filhas e ritmos diferentes de divisão são os responsáveis primários pela diferenciação em tecidos e órgãos de um organismo multicelular.
(b) Da MEIOSE, divisão especial que leva à formação das CÉLULAS GERMINATIVAS, as quais constituem as pontes biológicas entre as gerações.
As bases físicas da herança
A unidade da herança biológica é constituída por um segmento de uma
substância denominada ácido desoxiribonucleico (DNA). Onde se encontra esta última?
Nos cromossomos, elementos diferenciados que ocorrem nos núcleos de todas as
células. Análises de cromossomos isolados revelam dois componentes principais:
DNA e histona (uma proteína básica). Estas duas substâncias, em quantidades
aproximadamente iguais, constituem cerca de 90% da massa da maioria dos
cromossomos. Os restantes 10% são formados por proteína não-histônica, com uma
pequena porção de ácido ribonucleico (RNA).
Em termos muito simplificados, a síntese protéica ocorre da seguinte
maneira: o DNA, através do processo de transcrição, fornece um molde para o
RNA; e este, por sua vez, traduz esta informação para formar as proteínas. A
duplicação do DNA ocorre através de sua replicação. O esquema geral é o
seguinte:
Fig.1
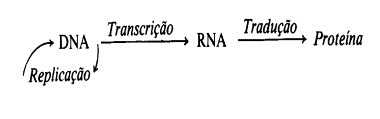
Quase todas as células podem sintetizar proteínas; o processo ocorre principalmente no citoplasma, mas também um pouco no núcleo. Os sítios de síntese são organelas chamadas ribossomos. Um segmento de DNA é denominado um gene. Quando ele ocorre em mais de uma forma (por exemplo, A e a), diz-se que apresenta diferentes alelos. Nos organismos com reprodução sexuada um certo indivíduo vai receber, com relação a uma determinada característica, um alelo de seu pai e réplicas do mesmo, ou outro, de sua mãe. Portanto, quando considerarmos um sítio genético determinado (loco), é sempre em termos de pares (e o organismo é denominado diplóide). Se os alelos são idênticos, diz-se que o indivíduo é homozigoto (por exemplo, AA ou aa); se diferentes, heterozigoto (Aa). O conjunto de todos os genes é denominado genótipo (ou genoma); e sua manifestação, fenótipo. Estes dois termos podem ser usados, também, com referência a um só sítio genético.
DOENÇAS HEREDITÁRIAS QUE AFETAM OS DENTES
HERANÇA LIGADA AO X. A HIPÓTESE DE LYON
Situação de consultório
Um senhor de 44 anos de idade procura-o para que faça a extração do
primeiro molar maxilar direito, bastante cariado. Informa-o, no entanto, de que
é hemofílico. Segundo ele, a queda dos dentes na primeira dentição ocorreu de
maneira normal, e apenas numa ocasião houve hemorragia de intensidade evidente.
Já extraiu quatro dentes da dentição definitiva, dois em cada intervenção. Na
primeira houve hemorragia evidente por 15 dias; na segunda, esta foi muito mais
acentuada, tendo ele de ser hospitalizado e receber uma transfusão. Nega a
ocorrência de gengivorragia. Segundo ele, três sobrinhos seus (dois filhos de
uma irmã com 30 anos e o terceiro de outra, com 28) também apresentam problemas
hemorrágicos. De comum acordo com o médico que trata desse senhor, você o
aconselha a uma dose profilática de crioprecipitado de Fator VIII,
transcorrendo a extração sem incidentes. Não é realizada a sutura do alvéolo
dentário, pois o fio atuaria como corpo estranho, aumentando o consumo de Fator
VIII. Posteriormente, foi-lhe aconselhada dieta líquida fria no primeiro dia,
líquida e pastosa no segundo e terceiro, e lavagem freqüente da boca com
solução isotônica de NaCl gelada, para evitar a formação de coágulo volumoso.
Aconselhou-se, também, que a higiene oral fosse feita com antissépticos.
A hemofilia é apenas uma das doenças hemorrágicas hereditárias com
manifestações orais. O seu padrão de herança típico, recessivo ligado ao X, já
era parcialmente conhecido no início da nossa era, como pode ser comprovado
pela leitura do Talmude.
Critérios para a identificação da herança
recessiva ligada ao X
1. O traço ocorre muito mais freqüentemente em
homens do que em mulheres.
2. Ele é passado de um homem afetado, através de
todas as suas filhas, para a metade dos filhos destas.
3. Não há nunca transmissão pai-filho.
4. O relacionamento entre irmandades com pessoas
afetadas se faz através de mulheres.
Critérios para a identificação da herança
dominante ligada ao X
1. Homens afetados terão todas as filhas afetadas, porém
nenhum filho.
2. A segregação da característica, na progênie de
mulheres afetadas, é indistingüível da observada na herança autossômica
dominante.
3. Observam-se mais mulheres que homens com a
condição (se a característica é rara, haverá duas vezes mais mulheres que
homens com a condição).
Herança holândrica
Genes localizados no cromossomo Y são herdados, como o sobrenome,
exclusivamente através da linguagem masculina. Um homem afetado transmitirá o
traço a todos os seus filhos, mas a nenhuma de suas filhas. Deve-se salientar,
no entanto, que existe apenas uma característica fenotípica simples
(hipertricose na orelha), que parece localizar-se neste cromossomo. Na maior
parte do mesmo ocorrem fatores relacionados com a espermatogênese, maturação dos
testículos e crescimento somático em geral. No que se refere especificamente à
dentição, o efeito ainda não bem caracterizado deste cromossomo parece ser o de
retardar a época de maturação dentária, mas também o de condicionar dentes
maiores no final do processo.
Herança limitada e influenciada pelo sexo
Existem genes localizados nos autossomos, cuja expressão, porém,
depende do sexo do indivíduo portador. Quando a dependência de expressão é
absoluta, isto é, quando o gene só se manifesta em um sexo, fala-se de herança
limitada ao sexo. Um exemplo é o de um gene que condiciona puberdade precoce em
homens (com aceleração do crescimento aos quatro anos de idade e fusão precoce
das epífises dos ossos longos) mas não apresenta efeito nenhum nas mulheres.
Por outro lado, certas características podem expressar-se nos dois
sexos, mas o fazem com muito mais freqüência em um do que no outro (herança
influenciada pelo sexo). O exemplo clássico, aqui, é a calvície, muito mais
comum em homens.
A hipótese de Lyon
As mulheres têm dois cromossomos X, e os homens um só. Entretanto,
produtos condicionados por genes localizados neste cromossomo são formados em
quantidade aproximadamente iguais. Como ocorre esta compensação de dose?
Supunha-se que, nos mamíferos, houvesse algum mecanismo regulatório que fizesse
com que a atividade, no cromossomo X isolado, fosse o dobro da atividade de
dois X quando estivessem juntos na mesma célula. Mary Lyon, uma pesquisadora
inglesa, foi a primeira a explicitar em detalhes uma teoria, hoje já amplamente
comprovada pelos fatos, fornecendo uma base física para o fenômeno. Em síntese,
a hipótese é a seguinte:
(a) Nas células somáticas das fêmeas dos mamíferos apenas um cromossomo
X é ativo. O segundo X está condensado e inativo, e aparece nas células
interfásicas como um corpúsculo bem delimitado localizado próximo à membrana
nuclear (a cromatina sexual).
(b) A inativação ocorre bem no início da vida embrionária.
(c) O X inativo pode ser de origem tanto paterna como materna, nas
diferentes células de uma fêmea. Mas, após a "decisão" de qual dos X
será inativado em uma determinada célula, todas as suas células descendentes
''mantêm a decisão", isto é, terão o mesmo X inativado. A inativação
ocorre ao acaso mas é fixa.
MANIFESTACÕES ORAIS E FACIAIS DAS
ANOMALIAS CITOGENÉTICAS
Situação de consultório
A mãe de uma criança (M.L.) com a síndrome de Down leva-a ao seu
consultório devido a doença periodontal séria. Trata-se de um menino de oito anos,
com aspectos característicos da doença: baixa estatura, hipotonia, retardo
mental, cabeça achatada, olhos com pregas epicânticas, mãos curtas e largas com
uma única linha palmar (prega simiesca) e clinodactilia (encurvamento) do
quinto dedo. O exame oral revela, além da doença periodontal (muito freqüente
nestes casos), língua fissurada e aumentada em relação ao normal. O volume da
mesma faz com que M.L. permaneça quase permanentemente de boca aberta. Os
caninos, os primeiro e segundo morares ainda são da dentição decídua, e
apresentam-se um pouco aumentados em relação ao tamanho normal, em contraste
com os incisivos centrais e laterais, bem como os primeiros molares
permanentes, que são de tamanho reduzido. Há ligeira assimetria nas arcadas.
Apesar da higiene precária, havia apenas um dente cariado.
A síndrome de Down, ou mongolismo, é causada por aberrações
cromossômicas que resultam na ocorrência, em dose tripla, de material do
cromossomo 21. É a mais comum das doenças originadas por problemas deste tipo.
Aberrações cromossômicas
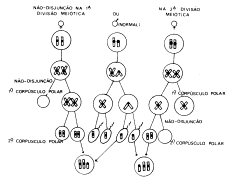
As anomalias
cromossômicas podem ser numéricas ou estruturais. As primeiras surgem
principalmente através do processo de não-disjunção (falha na separação de
cromossomos pareados ou cromátides irmãs na anáfase, tanto em uma divisão
mitótica como na primeira ou na segunda divisão meiótica. A Fig.2 representa a
não-disjunção do cromossomo x.
Fig.2 –
Formação de um indivíduo XXY por não-disjunção na primeira (esquerda) ou na
segunda (direita) divisão meiótica.
Se a não-disjunção ocorre em apenas um cromossomo, podem originar-se
células ou monossômicas (Fig. 3.i) ou trissômicas (Fig. 3.j). Se a alteração se
dá em dois cromossomos pode ocorrer tetrassomia (Fig. 3.k). Por outro lado, se
os problemas de separação envolverem o lote inteiro, podem formar-se células
haplóides, triplóides ou tetraplóides (Figs. 3.l, 3.m e 3.n, respectivamente).
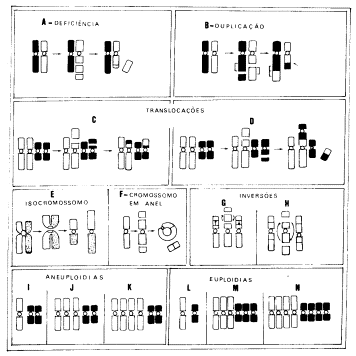
Fig. 3 – Esquema dos diferentes tipos de aberrações
cromossômicas
Nas alterações estruturais a modificação ocorre nos cromossomos devido
a quebras de natureza diversa. Alguns dos tipos que se formam são mostrados na
Fig.3 e são: deficiências (3.a), duplicações (3.b), translocações (3.c e 3.d),
isocromossomos (3.e), cromossomos em anel (3.f) e inversões (3.g e 3.h).
DOENÇAS CROMOSSÔMICAS
Além da síndrome de Down, cujas características foram mencionadas
acima, já existe um grande número de síndromes razoavelmente caracterizadas e
que são condicionadas por aberrações cromossômicas. Informações sobre algumas
das mesmas (as mais comuns) são apresentadas na tabela a seguir:
|
Sintomas gerais e manifestações orais
e faciais em algumas doenças cromossômicas |
||
|
Aberração |
Sintomas Gerais |
Manifestações orais e faciais |
|
45,X (Síndrome de Turner) |
Baixa estatura, amenorréia primária devido a
gônodas em fita, infantilismo sexual, mamas mais afastadas que o comum,
cúbito valgo, pescoço alado e linfedema das extremidades (em recém-nascidos). |
Micrognatia, dentes mal posicionados,
hipoplasia do maxilar, palato ogival. |
|
Mulheres poli-X |
Fenótipo geralmente não característico.
Cerca de 62% das pessoas com essas constituições (47, XXX; 48, XXXX; 49,
XXXXX) não apresentam anormalidades e 73% têm menstruação e desenvolvimento
dos selos normais. O risco de deficiência mental, no entanto, aumenta com o
número de cromossomos X presentes. |
Prognatismo relativo, lábios perfurados,
palato fendido. |
|
47, XXY e variantes (Síndrome de Klinefelter) |
Testículos hipoplásticos com hialinização
tubular, estatura elevada, ginecomastia, caracteres sexuais masculinos pouco
desenvolvidos. À medida que aumenta o número de cromossomos X, cresce o risco
de deficiência mental e agressividade. |
Prognatismo mandibular (XXYY), palato
fendido (15% de XXXXY), taurodontismo freqüente em XXXY e XXXXY. |
|
XYY |
Estatura alta, anormalidades esqueléticas
pouco pronunciadas, comportamento agressivo e anti-social. |
Prognatismo, palato estreito e alto, lesões
císticas na mandíbula, dentes grandes. |
|
5p- (Síndrome do miado do gato) |
Faringe hipoplástica, originando choro
característico, retardo psicomotor grave, hipotonia muscular, microcefalia. |
Micrognatia, úvula bífida. |
|
47, + 13 (Síndrome de Patau) |
Provavelmente a anormalidade cromossômica mais
grave compatível com vida extra-uterina. Retardo psicomotor severo,
microcefalia, muitas mal formações dos músculos e esqueleto, surdez, defeitos
cardíacos. |
Micrognatia (90%), lábio leporino com ou
sem palato fendido (80%), língua recortada. |
|
47, + 18 (Síndrome de Edwards) |
Hipertonia, ocipital proeminente, esterno
curto, problemas cardíacos, criptorquidismo, dedos sobrepostos com
deformidades de flexão. |
Micrognatia, boca pequena, lábio leporino e
palato fendido em 15% dos casos. |
|
47, + 22 |
Anomalias cardíacas e genitourinárias,
retardo mental grave, atresia anal, hipotonia grave, coloboma da íris. |
Hipertelorismo, orelhas mal formadas,
palato fendido, micrognatia. |
SÍNDROME DE TURNER (45,X e variantes)
Fig.4 – Fenótipo de uma mulher com a síndrome de
Turner 45,X.
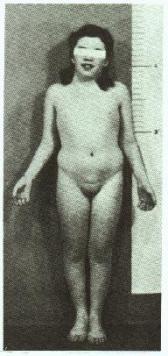
Ao contrário dos
pacientes com outras aneuploidias dos cromossomos sexuais, as meninas com a
síndrome de Turner freqüentemente são identificadas ao nascimento ou antes da
puberdade por suas características fenotípicas distintivas (Fig. 4).
A síndrome de Turner é bem menos comum do que outras aneuploidias dos
cromossomos sexuais. A incidência do fenótipo da síndrome é de cerca de 1 em
5.000 meninas nativivas. A constituição cromossômica mais constante é 45,X (às
vezes escrita, na literatura antiga, como 45,XO) sem um segundo cromossomo
sexual, X ou Y. Contudo, 50% dos casos possuem outros cariótipos. Um quarto dos
casos envolve cariótipos em mosaico, nos quais apenas uma parte das células é
45,X. Os cariótipos mais comuns e suas freqüências relativas são os seguintes
(Hook e Warburton, 1983):
|
45,X Mosaicos 45,X / 46,XX 46,X,i(Xq) Mosaicos 45,X / 46,X,i(Xq) Deleções 46,XXq- ou 46,XXp- Outros mosaicos 45,X/2 |
53% 15% 10% 8% 6% 8% |
A constituição cromossômica é clinicamente significativa; por exemplo,
as pacientes com um isoXq são semelhantes às clássicas pacientes 45,X, enquanto
as pacientes com uma deleção de Xp têm baixa estatura e malformações
congênitas, e aquelas com deleção de Xq freqüentemente apresentam apenas
disfunção gonadal.
As anormalidades típicas da síndrome de Turner abrangem baixa estatura,
disgenesia gonadal (de regra, gônadas vestigiais), fácies incomum típica,
pescoço alado, linha posterior de implantação dos cabelos baixa, tórax largo
com mamilos amplamente espaçados e uma freqüência elevada de anomalias renais e
cardiovasculares. Ao nascimento, os bebês com esta síndrome têm, muitas vezes,
edema do dorso do pé, um sinal diagnóstico útil. Muitas pacientes apresentam
coarctação da aorta. Linfedema pode estar presente na vida fetal, causando
higroma cístico (visível por ultra-sonografia), que é a causa do pescoço alado
observado após o nascimento. A inteligência costuma ser média ou acima da
média. Contudo, muitas pacientes exibem deficiência da percepção espacial,
organização motora perceptiva, ou execução motora refinada. Em conseqüência, o
QI não-verbal é bem mais baixo que o QI verbal.
A freqüência muito alta de 45,X em abortos espontâneos já foi
mencionada. Esta anormalidade é responsável por 18% dos abortos espontâneos
cromossomicamente anormais e está presente numa proporção estimada em 1,5% dos
conceptos. O único X geralmente é de origem materna; em outras palavras, o erro
meiótico costuma ser paterno. Desconhece-se o motivo da freqüência
extraordinariamente alta de não-disjunção do cromossomo X ou Y na meiose
paterna. Ademais, não está claro por que o cariótipo 45,X, tão letal in utero, parece ser completamente
compatível com a sobrevida pós-natal.
Na idade adulta, muitas pacientes com a síndrome de Turner se afligem
por sua infertilidade e baixa estatura. Embora a terapia com estrogênios possa
levar ao desenvolvimento dos órgãos genitais internos e externos, caracteres
sexuais secundários e menstruações, não corrige a infertilidade, que é uma
característica quase constante, resultado da atresia das células germinativas
iniciais. Atualmente se estuda o possível valor de baixas doses de estrogênio,
androgênio e hormônio do crescimento na terapia da baixa estatura na síndrome
de Turner. Até agora, poucos estudos envolvendo grandes números de pacientes
forneceram dados sobre o impacto destes agentes na estatura adulta final, mas
está claro que cada droga pode afetar a taxa de crescimento a curto prazo.
Embora a grande maioria das pacientes 45,X seja de mulheres
fenotípicas, muito raramente encontra-se um conjunto de cromosomos 45,X num
homem fenotípico que tem testículos mas é estéril. Os homens 45,X podem ter
iniciado a vida como mosaicos 45,X/46,XY, nos quais a linhagem XY se perdeu,
pelo menos no tecido estudado, ou podem ter uma translocação Y;autossomo não
reconhecida envolvendo o 1ócus do FDT. Há também alguns casos raros de mulheres
46,XY com estigmas da síndrome de Turner. Em todos estes casos, uma parte do cromossomo
Y está deletada (Levilliers et al., 1989; Fisher et al., 1990).
TRISSOMIA DO X (47,XXX)
A trissomia do X e as síndromes mais raras de tetrassomia do X
(48,XXXX) e pentassomia do X (49,XXXXX) são os equivalentes na mulher da
síndrome de Klinefelter masculina. As mulheres com trissomia do X, embora de
estatura geralmente acima da média, não são fenotipicamente anormais. Algumas
são identificadas em clínicas de infertilidade e outras em instituições para
retardados mentais, mas provavelmente muitas permanecem sem diagnóstico. Os
estudos de acompanhamento mostraram que as mulheres XXX sofrem as alterações da
puberdade numa idade apropriada, mas há relatos de puberdade precoce em certas
pacientes. Algumas deram à luz crianças, e estas são praticamente todas
cromossomicamente normais. Há um déficit significativo do desempenho em testes
de QI, e cerca de 70% dos pacientes têm problemas do aprendizado graves.
Nas células 47,XXX, dois dos cromossomos X são inativados e de
replicação tardia, o que foi sugerido originalmente pelo achado de dois
corpúsculos de Barr. Quase todos os casos resultam de erros na meiose materna,
sendo a maioria na meiose I. Há um efeito da idade materna avançada, restrito
aos pacientes nos quais o erro ocorreu na meiose I materna (May et al., 1990).
A síndrome de tetrassomia do X está associada a atraso mais grave do
desenvolvimento físico e mental, e a síndrome de pentassomia do X, assim como o
XXXXY, geralmente inclui grande retardo do desenvolvimento com múltiplos
defeitos físicos que lembram a síndrome de Down.
SÍNDROME DE KLINEFELTER (47,XXY)
A Fig.5 mostra o fenótipo da síndrome de Klinefelter. Os pacientes são
altos e magros, com membros inferiores relativamente longos. Parecem
fisicamente normais até a puberdade, quando os sinais de hipogonadismo se
tornam óbvios. Os testículos permanecem pequenos e os caracteres sexuais
secundários continuam subdesenvolvidos. Os pacientes Klinefelter quase sempre
são inférteis.
A incidência é de cerca de 1 em 1.000 meninos nativivos (1 em 2.000
nascimentos totais) e 1 em 300 abortos espontâneos. Embora o fenótipo pareça
benigno em comparação com o das trissomias autossômicas, metade das concepções
47,XXY perde-se antes do nascimento.
Fig.5 –
Fenótipo de um homem com a síndrome de Klinefelter 47,XXY. Observe os membros
longos e órgão genitais relativamente pequenos. A ginecomastia, observada neste
paciente, não é uma manifestação constante.
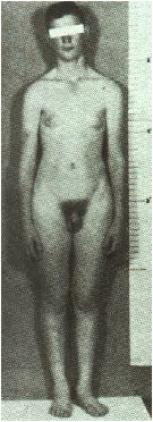
Cerca de 15% dos
pacientes Klinefelter têm cariótipos em mosaico. Como grupo, esses pacientes
mosaicos apresentam fenótipos variáveis; alguns têm desenvolvimento testicular
normal. O cariótipo em mosaico mais comum é 46,XY/47,XXY, provavelmente em
conseqüência da perda de um cromossomo X num concepto XXY durante uma divisão
pós-zigótica inicial. Conforme previsto pelo achado de que os pacientes
Klinefelter 47,XXY têm um corpúsculo de Barr, um dos dois cromossomos X é
inativado.
Numa pesquisa citogenética e molecular combinada da origem parental e
do estágio meiótico do erro não-disjuncional responsável pela síndrome,
verificou-se que metade dos casos resulta de erros na meiose I paterna, um
terço de erros na meiose I materna e os demais de erros na meiose II ou de um
erro mitótico pós-zigótico levando a mosaicismo. A idade da mãe é elevada nos
casos associados a erros na meiose I materna, mas não nos outros casos (Jacobs
et al., 1988).
Embora haja ampla variação fenotípica entre os pacientes com esta e
outras aneuploidias dos cromossomos sexuais, exatamente como na população em
geral, identificaram-se algumas diferenças fenotípicas constantes entre os
pacientes com a síndrome de Klinefelter e homens cromossomicamente normais.
Seus escores em certos testes de desempenho da inteligência (testes de QI) é
pouco - mas significativamente - reduzido. Dois terços dos pacientes apresentam
problemas educacionais, especialmente dislexia, enquanto menos de um quarto das
pessoas normais tem dificuldades de aprendizado. A puberdade ocorre na idade
normal, mas o tamanho testicular permanece bem abaixo da média. A ginecomastia,
enfatizada como um achado típico na literatura antiga, em geral está ausente.
Muitos dos meninos afetados mostram uma adaptação psicossocial relativamente
fraca.
Existem diversas variantes da síndrome de Klinefelter, com outro
cariótipo que não 47,XXY, incluindo 48,XXYY, 48,XXXY e 49,XXXXY. Como regra, os
cromossomos X adicionais causam um cariótipo correspondentemente mais anormal
(embora os cromossomos X extras sejam inativos), com maior grau de dismorfismo,
desenvolvimento sexual mais deficiente e debilitação mental mais intensa. Uma
observação inesperada nos pacientes 49,XXXXY (e em seus equivalentes femininos,
com cariótipos 49,XXXXX) é ser o fenótipo semelhante, em muitos aspectos, ao da
síndrome de Down. Esta observação depõe contra o concerto amplamente difundido
de que o fenótipo da síndrome de Down depende estritamente da dosagem tripla
dos genes no cromossomo 21 e, em seu lugar, sugere um atraso mais geral no
desenvolvimento relacionado a um desequilíbrio cromossômico.
SÍNDROME 47,XYY
Apesar de a constituição cromossômica 47,XYY não estar associada a
nenhum fenótipo obviamente anormal, ela despertou grande interesse médico e
científico após observar-se que a proporção de homens XYY era bem maior entre
os detentos de uma prisão de segurança máxima, sobretudo entre os mais altos,
do que na população em geral (Jacobs et al., 1968). Cerca de 3% dos homens em
penitenciárias e hospitais de doentes mentais possuem um cariótipo 47,XYY; no
grupo com altura acima de 1,80 m, a incidência é bem maior (mais de 20%).
Dentre os meninos nativivos, a freqüência do cariótipo 47,XYY é de cerca de 1
em 1.000.
A origem do erro que leva ao cariótipo XYY deve ser a não-disjunção
paterna na meiose II, produzindo espermatozóides YY. As variantes XXYY e XXXYY
menos comuns, que compartilham as manifestações das síndromes XYY e de
Klinefelter, provavelmente também se originam do pai, numa seqüência de eventos
não-disjuncionais nas meioses I e II.
Os meninos XYY identificados em programas de triagem neonatal sem vício
de averiguação são altos e têm um risco aumentado de problemas do
comportamento, em comparação com meninos cromossomicamente normais. Possuem
inteligência normal e não são dismórficos. A fertilidade é regular e parece não
haver nenhum risco aumentado de que um homem 47,XYY tenha um filho com
cromossomos anormais. Muitos pais de crianças identificadas, antes ou após o
nascimento, como XYY, tornam-se extremamente preocupados com as implicações
comportamentais. Alguns médicos acreditam que a informação deve ser omitida
quando a identificação é feita após o nascimento. A incapacidade de avaliar o
prognóstico em cada caso torna a identificação de um feto XYY um dos problemas
de informação genética mais sérios enfrentados em programas de diagnóstico
pré-natal.
SÍNDROME DO MIADO DE GATO (5p-)
Fig.6 – Lactente com a
síndrome do miado de gato, que resulta de deleção de parte do cromossomo 5p.
Observe a fácies com hipertelurismo, epicanto e retrognatia.
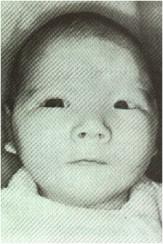
A síndrome do miado de
gato, na qual há uma deleção do braço curto do cromossomo 5, recebeu sua
denominação porque o choro de um lactente afetado assemelha-se ao miado de um
gato. É responsável por 1% dos pacientes com retardamento mental
institucionalizados. A aparência facial, vista na Fig.6, é distintiva, com
microcefalia, hipertelorismo, inclinação antimongolóide das fissuras
palpebrais, pregas epicânticas, orelhas de implantação baixa, às vezes com
apêndices pré-auriculares, e micrognatia.
A maioria dos casos da síndrome do miado de gato é esporádica, mas l0 a
15% dos pacientes são filhos de portadores de translocação. A Fig.7 mostra um
heredograma ilustrativo.
Fig.7 –
Heredograma de uma criança com a síndrome do miado de gato, cuja mãe era
portadora de uma translocação balanceada entre os cromossomos 5p e 9p. A mãe
teve (1) a criança afetada, que recebeu seu cromossomo 9 normal, (2) uma filha
retardada, que recebeu seu cromossomo 5 normal e o cromossomo 9 translocado (e
portanto era trissômica para o segmento translocado do cromossomo 5p), (3) uma
filha portadora da translocação balanceada e, após amniocentese e análise
cromossômica, dois filhos do sexo masculino: (4) um portador da translocação
balanceada e (5) um com cariótipo normal.
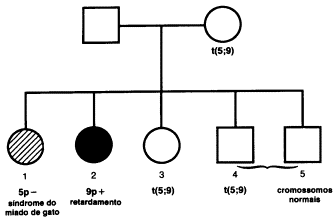
Os pontos de quebra e
a extensão do segmento deletado do cromossomo 5p variam nos pacientes, mas a
região crítica, ausente em todos os pacientes com o fenótipo, foi identificada
como a banda cromossômica 5p15. Demonstrou-se que várias sondas de DNA e genes
estão deletados dos cromossomos 5p-. Contudo, resta explicar a causa da relação
entre monossomia para tais genes e o fenótipo clínico.
ODONTOLOGIA RACIAL
Genética de populações
Genética de populações é o ramo da genética que trata dos problemas
relacionados com a distribuição dos genes e genótipos nas populações.
Preocupa-se, portanto, com a natureza e origem das diferenças genéticas, sua
manutenção ou processo de eliminação, bem como com o cálculo de suas
freqüências relativas.
Um odontólogo precisa ter noções sobre esta área da genética, uma vez
que, através desta informação, ele poderá calcular a freqüência de portadores
assintomáticos de doenças hereditárias que afetam os dentes, o que é muitas
vezes indispensável para fornecer aconselhamento sobre probabilidade de prole
afetada. Há, porém, uma área de atuação do odontólogo na qual é especialmente
importante ter noções claras sobre a genética de populações: é a do uso dos
raios-X para fins diagnósticos de problemas dentários.
A genética de populações tem como seu princípio básico a lei de
Hardy-Weinberg, assim denominada em homenagem a seus descobridores, G. H.
Hardy, matemático inglês, e W. Weinberg, médico alemão. Antes da derivação
desta lei, em 1908, partindo da observação de que nas segregações familiares a
proporção, no casamento de dois heterozigotos para um par de genes
autossômicos, era de 3 fenótipos dominantes para 1 recessivo, supunha-se que
fatalmente, após um determinado número de gerações, só deveriam subsistir na
população genes dominantes (uma situação que poderia ser denominada de
genofagia). Estes dois cientistas, no entanto, demonstraram que na ausência de fatores que perturbem esta
distribuição e numa população com cruzamentos ao acaso, a proporção de
caracteres dominantes e recessivos pode ser de qualquer tipo e as freqüências
relativas de cada alelo tendem a permanecer constantes de geração a geração.
Simbolizando-se por p a freqüência do
alelo A e por q a de a, pode-se demonstrar
facilmente que a distribuição genotípica, na população, será de p²AA + 2pq Aa + q²aa.
Se não há alterações de freqüências gênicas entre gerações, isto
significa que vivemos em um mundo imutável, cada geração monotonamente
repetindo o que ocorreu na anterior? Não, a lei de Hardy-Weinberg descreve
somente a estática da distribuição gênica, e é válida apenas para genes em
equilíbrio. Os principais fatores que podem alterar este equilíbrio são a
mutação e a seleção natural. Em casos especiais, e principalmente em populações
pequenas, a deriva genética também pode ser responsável por mudanças.
Mutação pode ser considerada como qualquer alteração no material
genético. Essas alterações ocorrem espontaneamente em uma taxa média de
1:100.000 ( lx10 -5); isto é, em cada geração celular, em um loco
especifico, 1 alelo em cada 100.000 replica-se erroneamente.
Porém, existem fatores que podem aumentar essa taxa de mutação. Tanto
as radiações, como substâncias químicas diversas, podem aumentar a taxa
espontânea de mutação. A unidade de dose de radiação absorvida é denominada
rad. A radiação natural talvez forneça 130 milirads de exposição geneticamente
efetiva (isto é, que alcança as gônadas) por ano, enquanto a radiação produzida
pelo homem (especialmente a utilizada para fins diagnósticos na medicina e
odontologia) adiciona mais cerca de 60 milirads (total: 190). Calculando-se em
30 anos o período reprodutivo, ter-se-ia uma exposição total de talvez 6 rads.
Quais seriam os efeitos desta exposição? Há dois princípios básicos de
radiogenética que devem ser sempre lembrados:
(a) quanto a efeitos genéticos, não há limiar, isto é, qualquer dose,
por pequena que seja, é potencialmente mutagênica;
(b) as doses são cumulativas.
Isto significa que deve ser evitada qualquer irradiação desnecessária.
A propósito, recorde-se que uma radiografia de toda a arcada dentária fornece
uma dose, à superfície do corpo, de 5r! E que, como as mutações ocorrem ao
acaso, dificilmente elas serão benéficas, envolvendo quase sempre mudanças
deletérias.
Também não deve-se confundir seleção natural com deriva genética. A
definição de Charles Darwin de seleção natural é muito simples e clara; ela
envolve "a preservação das diferenças individuais e variações favoráveis e
a destruição das prejudiciais''. Constitui este processo, juntamente com a
mutação, os dois fatores fundamentais responsáveis pela evolução biológica. A
seleção determina mudanças de cunho determinista (podem ser previstas); mas há
elementos de puro acaso que podem influir na passagem dos genes de uma geração
a outra, que são englobados sob a denominação geral de deriva genética.
Variação racial de características
dentárias
Os processos acima indicados são responsáveis pela variabilidade
genética existente numa espécie; através do isolamento geográfico é possível o
estabelecimento, dentro de cada espécie, de unidades populacionais
características, as raças.
Pode-se definir raça, para a espécie humana, como o “conjunto de
indivíduos geográfica ou culturalmente mais ou menos isolados, que diferem
geneticamente de outros grupos similares”. Examinada dentro deste contexto esta
unidade populacional pode ser considerada a resultante inicial do processo de
diversificação evolutiva.
As raças diferem entre si em um grande número de características
dentárias. Algumas dessas diferenças relacionam-se com a seqüência e cronologia
da erupção dentária, bem como com o tamanho e proporção de dentes e arcadas,
aspectos já abordados anteriormente neste livro. Duas das características mais
estudadas, que serão aqui examinadas, são os incisivos em forma de pá e o
tubérculo de Carabelli.
Incisivos em forma de pá
Fig.8 – Os
quatro tipos de incisivos em forma de pá estabelecidos por Hrdlička. Há uma gradação, desde uma manifestação marcante (a), até a
ausência da característica (d).
Quando, nesses dentes,
há na superfície lingual margens laterais proeminentes, criando uma depressão,
diz-se que eles têm a forma de uma pá. Há uma gradação na manifestação da
característica, e o antropólogo norte-americano A. Hrdlička classificou-a de acordo com quatro tipos
(Fig.8). Há, no entanto, formas intermediárias. A herança é do tipo poligênico,
mostrando acentuada variação racial. Enquanto em brancos ocorrem incisivos
moderada ou marcantemente com esta forma em apenas 10% das pessoas examinadas,
esta freqüência eleva-se a mais de 70% em chineses e japoneses, podendo
alcançar valores de até l00% em índios americanos e esquimós.
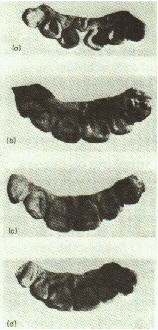
Tubérculo de Carabelli
Consiste numa alteração morfológica normalmente localizada na
superfície palatal da cúspide mesio-lingual dos molares maxilares permanentes
ou decíduos. A variação inclui desde uma pequena depressão, apenas, até a
presença de um tubérculo grande (Fig.9). O dente mais comumente afetado é o
primeiro molar. O tipo de herança desta característica também não está bem
definido; provavelmente mais que um par de genes influi na sua manifestação,
assim como fatores ambientais. Grupos asiáticos apresentam baixa freqüência
deste traço (25-60%), enquanto europeus e africanos mostram altas prevalências
(70-90%).
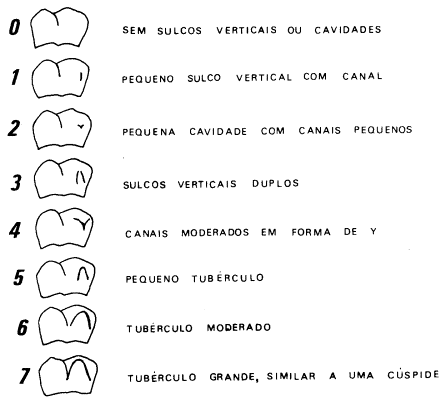
Fig.9 – Classificação em 8 tipos, do tubérculo de
Carabelli
AÇÃO GÊNICA
Os termos dominante e recessivo foram usados pela primeira vez por G. Mendel, o criador da Genética, e já fazem parte do nosso vocabulário diário. Um gene dominante pode ser definido como aquele que se manifesta em dose simples; recessivos seriam aqueles só detectáveis em dose dupla. É claro que essas definições relacionam-se com o método de observação. Ao nível molecular todos os genes são dominantes, pois o estudo apropriado de seu produto primário inevitavelmente indicará qualquer alteração que tenha ocorrido no mesmo. Estritamente falando é o caráter (expressão fenotípica do gene), e não este último, que é dominante ou recessivo, embora os termos gene dominante ou gene recessivo sejam comumente usados.
Fatores ambientais podem influir na manifestação de uma característica. Se um alelo determinado dá origem a fenótipos diversificados diz-se que ele possui expressividade variada. Em situações extremas o alelo pode estar presente e não se manifestar fenotipicamente - nesses casos, fala-se de penetrância incompleta. Cada alelo tem apenas um efeito primário ao nível molecular. Porém, ele pode produzir uma série de outros efeitos secundários, dependendo do grau de participação deste produto primário em cadeias metabólicas diferentes. Denomina-se pleiotropia a propriedade de um alelo ou par de alelos produzirem efeitos fenotípicos múltiplos.
Os dados familiares relativos a uma determinada condição podem ser resumidos em árvores genealógicas ou heredogramas, utilizando-se os símbolos indicados na fig.10:
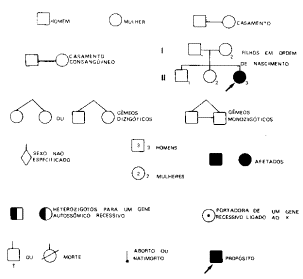
Fig.10
CRITÉRIOS
PARA A IDENTIFICAÇÃO DA HERANÇA AUTOSSÔMICA DOMINANTE
Há algumas regras simples que permitem, no caso de características hereditárias raras, identificar quais são autossômicas dominantes. Supõe-se sempre, nesses casos, expressividade constante e penetrância completa. As regras podem ser enumeradas do seguinte modo:
1) O traço aparece em gerações sucessivas, sem "pular" nenhuma;
2) Ele é transmitido de uma pessoa afetada a cerca da metade de seus descendentes;
3) Não há preferência sexual, isto é, a probabilidade de homens ou mulheres apresentarem a característica é a mesma, sendo idêntica, também, a probabilidade de transmiti-la.
CRITÉRIOS PARA A IDENTIFICAÇÃO DA HERANÇA AUTOSSÔMICA RECESSIVA
1) O traço aparece em irmãos, com genitores normais.
2) Em média, 1/4 dos irmãos do propósito (indivíduo afetado através do qual a família é localizada) possuem a característica.
3) Há um aumento na probabilidade de que os genitores sejam consangüíneos (porque, se a característica é rara, seria pouco provável o casamento de dois heterozigotos não relacionados biologicamente; no caso de eles serem primos, no entanto, pode-se explicar essa união rara pelo fato de ambos serem portadores de réplicas de um mesmo gene mutante, presente em um dos ancestrais comuns).
4) Também aqui não há preferência sexual na manifestação ou transmissão da característica.
CO-DOMINÂNCIA E HERANÇA INTERMEDIÁRIA
Se ambos os alelos de um par expressam-se de maneira completa no heterozigoto, diz-se que eles são co-dominantes. Por exemplo, uma pessoa do grupo sangüíneo AB apresenta tanto o antígeno A como o B em suas hemácias, substâncias essas formadas através da ação dos genes IA e IB, presentes em suas células. Se a expressão fenotípica do heterozigoto, contudo, ao invés de manifestar-se como um mosaico, mostra-se quantitativamente intermediária, diz-se que a herança é deste tipo (intermediária). Muitos dos erros inatos do metabolismo podem ser classificados como tendo este modo de herança, pois o doente (homozigoto para o alelo mutante) apresenta níveis mínimos de atividade enzimática, o homozigoto para o alelo normal atividade de 100% e o heterozigoto, em média, 50%. Exemplo: galactosemia, doença caracterizada por hepatoesplenomegalia, cirrose do fígado, cataratas e retardo mental, cansada por uma forma inativa da enzima galactose-1-fosfato uridil transferase.
Análise
Genealógica
1) Um menino de 4 anos tem os dentes pigmentados de marrom, pouco esmalte e dentina translúcida, permitindo visualizar-se os contornos da polpa. Mostra, ainda, desgaste das coroas e exposição de algumas polpas dentárias. Não há presença de cáries. Este quadro caracteriza a displasia da dentina e do esmalte. O heredograma da fig.11 mostra a distribuição de indivíduos afetados na família do propósito.
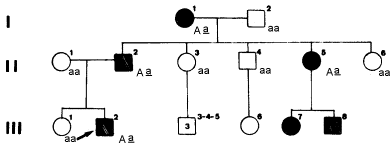
Fig.11 – Genealogia com diversos casos de displasia da dentina e do
esmalte.
Observando os critérios de identificação (a doença não pulou nenhuma geração, não há preferência sexual) percebe-se que o padrão de herança da anomalia é autossômico dominante. A partir daí é até possível determinar o genótipo do probando e dependendo do seu cônjuge a probabilidade de nascimentos com indivíduos afetados.
2) Na genealogia da fig.12 está segregando o gene que determina um tipo de raquitismo (dependente da vitamina D). O paciente apresenta, entre outros problemas, hipoplasia do esmalte, que se mostra na Segunda dentição opaco e amarelo-acinzentado, bem como grandes câmaras pulpares, tendo havido fechamento tardio dos ápices radiculares.
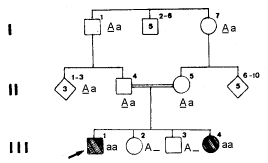 .
.
Fig.12 – Genealogia em que está segregando um gene responsável por
raquitismo dependente da vitamina D.
O padrão desta herança é autossômico recessivo. O heredograma permite visualizar a importância do casamento consangüíneo no aumento das manifestações de anomalias.
Determinando o genótipo dos indivíduos é fácil dizer que há 25% de chance de um novo irmão do probando ser afetado.
|
|
A |
a |
A |
AA |
Aa |
|
a |
Aa |
aa |
3) Fez-se a descrição de uma família residente em Porto Alegre, na qual cinco pessoas apresentavam a displasia fibrosa das maxilas conhecida como querubinismo (a fig.13 mostra dois deles). A informação obtida (fig14) inclui sete gerações, num total de 75 indivíduos. Vinte e três foram examinados por geneticistas e uma referência sobre pessoa já falecida foi confirmada através de fotografia. A afecção nessa família provavelmente é condicionada por um gene autossômico dominante com expressividade variável.

Fig.13 – Dois irmãos afetados por querubinismo (VI-11 e VI-13 na genealogia da Fig.14).
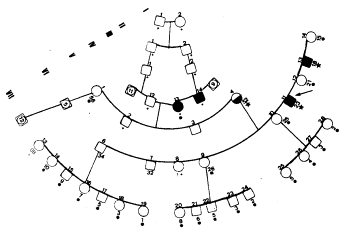
Fig.14 – Genealogia
da família adaptada ao lay-out da página com diversos casos de querubinismo.
Extraído do artigo: F.M. Salzano e H. Ebling. 1966. “Cherubism in a
Brazilian Kindred”. Acta Geneticae Medicae et Gemellologiae, Volume 15,
número 3, p.296-301.
CONCLUSÃO
Engenharia genética
Denomina-se engenharia genética a área de estudos surgida com os avanços espetaculares, ocorridos nos últimos anos, relacionados com a síntese, análise, transposição e manipulação em geral do DNA. Três fatores principais contribuíram para essas conquistas:
(a) a descoberta de maneiras de clivar o DNA em sítios bem específicos;
(b) o desenvolvimento de métodos simples e geralmente aplicáveis para a reunião de moléculas de DNA;
(c) a descoberta de técnicas efetivas para a introdução do DNA em organismos previamente refratários.
Quais são as aplicações possíveis da engenharia genética? Em primeiro lugar, devem ser considerados os aspectos puramente científicos. Por exemplo, a estrutura e organização do genoma de organismos superiores (como o homem) está sendo estudada intensivamente através desse enfoque. Esta pesquisa tinha sido, até pouco tempo, prejudicada pela complexidade desses genomas e a dificuldade de isolar-se porções particulares para a análise experimental. A inserção desses fragmentos em bactérias possibilitou a pesquisa de uma série de questões que só poderiam ser abordadas em sistemas desse tipo. A importância dessas investigações pode ser avaliada pelo fato de o Prêmio Nobel de Medicina, em 1978, Ter sido conferido a W. Arber, D. Nathans e H. O. Smith, justamente por seus estudos nas chamadas enzimas de restrição, capazes de fragmentar o DNA.
Em segundo lugar, é muito provável que em futuro próximo genes apropriados possam ser introduzidos em bactérias para convertê-las em fábricas bioquímicas para a produção de substâncias complexas de importância médica ou veterinária. Os exemplos aqui seriam os da insulina (para a qual parece iminente uma época de escassez), o hormônio do crescimento, anticorpos específicos para confeccionar vacinas contra a malária, febre aftosa e outras doenças infecciosas, e o fator VIII da coagulação sangüínea, necessário para os hemofílicos.
Outras aplicações incluem a transferência de genes para que plantas atualmente incapazes possam fixar o nitrogênio, o que seria muito importante para a agricultura.
Uma possibilidade mais remota seria a da alteração genética dirigida de nossa espécie, através de manipulações no DNA. Embora os meios para isso pareçam estar muito afastados, essa questão deve ser cogitada, pois criaria problemas éticos e morais muito importantes.
No entanto, essas pesquisas têm o seu perigo. Isso porque estão sendo criados organismos novos, autopropagáveis, para os quais talvez não haja possibilidade de defesa biológica por parte de nossa espécie. Foram estabelecidas, portanto, uma série de regras e regulamentos de segurança, para evitar a liberação involuntária de tais linhagens fora dos laboratórios.
As potencialidades biotecnológicas da engenharia genética condicionaram a formação de uma série de empresas, nos Estados Unidos e na Europa, e o interesse das multinacionais na sua aplicação comercial. E há pouco tempo a Suprema Corte dos Estados Unidos decidiu que podem ser patenteadas formas de vida criadas em laboratórios. Por 5 votos contra 4 a referida Corte deu ganho de causa a um processo da General Electric pedindo o direito de patentear um novo tipo de bactéria. O presidente da Corte na época, Warren E. Burger, justificou a decisão dizendo que não se tratava de uma lei ou coisa natural, propriedades de todos, mas de uma nova forma que surgiu graças à interferência humana, e portanto apresentando as características de um produto, como "o telefone, o transistor, o avião e a lâmpada elétrica''. Em todo caso, o juiz salientou que o Congresso poderia ser pressionado, pelos que têm outra opinião, a criar leis específicas visando as atividades da engenharia genética.
Eugenia e eufenia
A eugenia pode ser definida como aquele ramo do conhecimento que procura melhorar a qualidade genética de populações inteiras ao longo do tempo. Tradicionalmente, procuram-se distinguir dois objetivos dentro deste contexto geral:
(a) a redução na freqüência de genes presumivelmente deletérios (eugenia negativa); e
(b) a melhoria da constituição genética da população (eugenia positiva), embora o desenvolvimento de atividades relacionadas com uma das duas áreas de atuação implique necessariamente repercussões na outra.
Por outro lado, pode-se definir como eufênicas as medidas que procuram melhorar a ação gênica, seja pela eliminação de efeitos prejudiciais, seja pelo estímulo à produção de material útil.
Com a possibilidade de controle cada vez maior do processo de reprodução humana, cabe perguntar, como o fez Lappé (1975): pode uma política eugênica ser justa? Sua resposta é um sim qualificado, classificando ele de justa uma ação que prometesse melhoria real ou potencial em fatores sociais significativos para beneficiar um número máximo de pessoas, e que compensasse aqueles que fossem de alguma maneira privados por sua implementação. Já Beiguelman (1979), após fazer severas críticas aos movimentos eugenistas, mostrou-se cético quanto às vantagens que poderiam advir de ações neste sentido.
As restrições quanto à eufenia centram-se em seu possível aspecto disgênico. Com efeito, quando se corrige a ação de um gene deletério, fazendo com que seu portador tenha uma vida praticamente normal, automaticamente se está favorecendo a manutenção e reprodução deste gene prejudicial. Mas, seria difícil encontrar argumentos contrários à melhoria geral das condições ambientais das populações humanas, de maneira a maximizar a expressão de suas características genéticas positivas. Neste caso, a simples eliminação da pobreza é um objetivo ético que deveria ser perseguido por todos.
GLOSSÁRIO
• Atresia
= imperfuração dos orifícios naturais do corpo; estreitamento ou estenose dos
órgãos ocos.